
circular RNA expression profiling by RNA-seq faces major numerical and practical challenges. Numerically, circular RNAs as a population are typically present at much lower abundance levels, at about 5~10% of linear RNAs (Fig. 1A). The cross circular junction sequences necessary for circular RNA identification are even lower as a fraction of the full length transcripts (Fig.1B). In fact, in the circular RNA studies based on large data sets and deep coverages, circular junction sequencing reads are detected by only a few counts in the samples (Fig. 1C) [1].
Although a few observed circular junction reads provide the confidence of detecting the presence of a circular RNA, such low count numbers are below the limit of quantification. For differential expression analysis, at least several hundred read counts are needed to achieve errors < 20% (Fig. 2A) [2]. At a typical RNA-seq coverage of 30 million reads, less than 5% of the circular RNAs are distributed in the reliable range (Fig. 2B). In other words, > 95% circular RNAs are not quantified accurately. On the other hand, microarray accuracy is relatively unaffected by the lowered transcript levels (Fig. 2C) [3], which is well suited for circular RNAs.
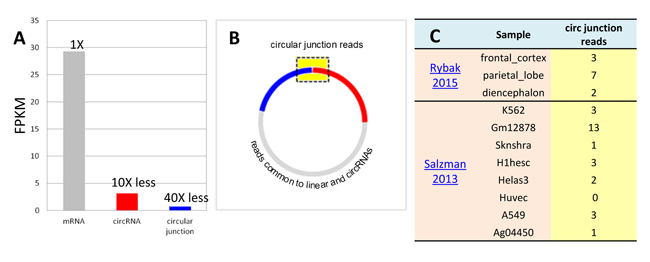
Figure 1. (A) Compared with mRNAs, circular RNA and circular RNA junctions as a population are at low abundance. (B) Circular junction reads only constitutes a fraction of the entire transcript. (C) A random example of hsa_circ_0054254 read counts across the tissue samples. Rybak2015, Salzman2013
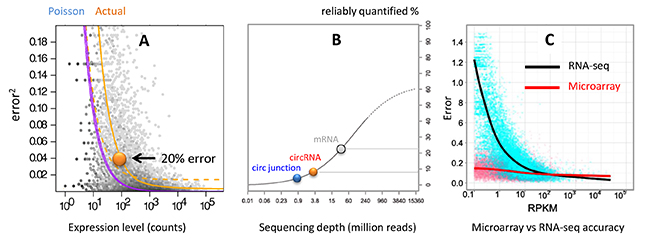
Figure 2. (A) Theoretical Poisson and Actual relationships between read count and quantification error [2]. (B) Sequencing depth at which the percentage of transcript population can be reliably quantified [4]. mRNA, circRNA and circular junction populations are indicated by the colored spheres at a RNA-seq depth of 30 million. (C) Quantification error vs transcript level by RNA-seq and microarray. Microarray accuracy is less affected by the transcript level [3].
In practice, generic RNA-seq, mostly intended for mRNAs, are inadequate or simply unavailable as a provided service for circular RNA profiling (Table 1). circRNA sequencing data analysis requires specialized databases, de novo transcript assembly, read mapping, and algorithms, all of which contribute to the analysis complexity, reduced reliability and accuracy, and the generally availability [5].
Arraystar circRNA Arrays use unique circular junction specific probes, combined with enzymatic linear RNA removal, to enrich, capture and quantify circular RNA targets at high sensitivity and specificity. As microarray hybridization signal is relatively unaffected by the presence of other high abundance RNAs, high sensitivity at one transcript per cell can be achieved. Overall, the circRNA arrays are much more an efficient, robust, and accurate technology that outperforms RNA-seq in circRNA profiling (Table 1). With the outstanding circular RNA contents, analyses and annotation, Arraystar circRNA Arrays are the only practical choice for circRNA profiling for biologists and clinicians.
Table 1.
| Arraystar circRNA microarrays | Sequencing |
| High sensitivity at as low as 1 transcript/cell | Most circRNAs cannot be quantified due to low circular junction read counts Deep sequencing needed at high costs |
| Unambiguous, reliable circular junction-specific array probes | Less reliable, less accurate, and less mature computational methods for circular RNAs |
| Dedicated circRNA annotation and analysis to gain insights into the circRNA biology, e.g. miRNA binding sites, miRSVR scores and conservation status, to unravel functional roles as miRNA sponges | Limited annotation information from the public circular RNA databases that are typically used by circular RNA sequencing suppliers |
| Desktop computational resources | High demand of computing power and ~1000X larger data storage |
Related Services
Circular RNA Array Service
Circular RNA qPCR Service
References
1. Salzman, J., et al. (2013) Cell-type specific features of circular RNA expression. PLoS Genet. 9(9):e1003777 [PMID: 24039610]
2. Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11(10):R106 [PMID: 20979621]
3. Xu, W., et al. (2011) Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A. 108(9):3707-12 [PMID: 21317363]
4. Labaj, P. P., et al. (2011) Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics. 27(13):i383-91 [PMID: 21685096]
5. Szabo, L. and Salzman, J. (2016) Detecting circular RNAs: bioinformatic and experimental challenges. Nat Rev Genet. 17(11):679-692 [PMID: 27739534]