GST-YTH is a recombinant fusion protein of YTH-DF2 m6A reader domain (a.a385-579) with a GST tag for m6A RNA enrichment. YTH is an evolutionarily conserved structural domain that selectively “reads” and binds m6A within the consensus RRACH motifs [1]. Structurally, the YTH domain contains two or three tryptophan residues that form an aromatic cage and binding pocket for m6A, with additional interactions with nucleotides before and after m6A, thus giving sequence preference to the RRACH motif [2-4](Fig.1). That is, GST-YTH binds m6A-containing RNAs in an m6A structure- and RRACH sequence motif-dependent manner [1], both of which are distinct from m6Am and other similar RNA modifications. Thus, GST-YTH pulldown is highly specific to m6A without the cross-reactivity with other structurally similar RNA modifications[5], particularly m6Am [6], as by m6A antibody MeRIP.
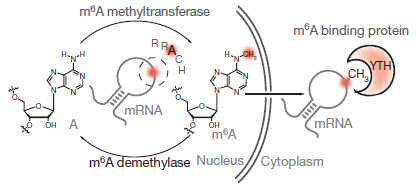
Figure 1. YTH binds to m6A-modified RNAs in an m6A structure- and RRACH sequence motif-dependent manner, at higher specificity to m6A without the cross-reactivity to other similar RNA modifications such as m6Am.
Reference
1. Wang X et al: N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505(7481):117-120.[PMID: 24284625]
2. Luo S, Tong L: Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc Natl Acad Sci U S A 2014, 111(38):13834-13839.[PMID: 25201973]
3. Theler D et al: Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res 2014, 42(22):13911-13919.[PMID: 25389274]
4. Xu C et al: Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol 2014, 10(11):927-929.[PMID: 25242552]
5. Linder B et al: Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods 2015, 12(8):767-772.[PMID: 26121403]
6. https://sysy.com/product/202003
Quantifying the percentage of modification for each transcript by microarray
The modified and unmodified fractions of the same RNA transcript, which differ only in their structures or the readers that bind to them, can assume different fates [4] (Figure 1). Importantly, the percentage of modified transcripts is highly relevant to their functional consequences. While current RNA modification profiling methods, such as MeRIP-seq (i.e. m6A-seq), can map the modification locations, they do not quantify the relative fraction of modified and unmodified RNA for a given transcript. Arraystar Epitranscriptomic Microarrays have the power to determine the percentage of modified transcripts by measuring the percentage of modification for each transcript in two color channels on the same array (Figure 2), while simultaneously profile what gene transcripts are modified and the differential modification between conditions.
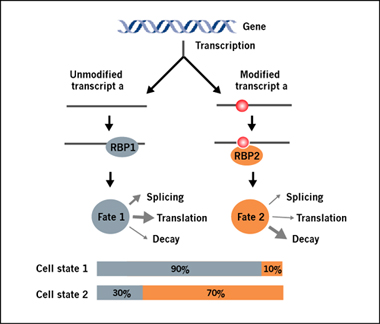
Fig 1. The changing modification stoichiometry generates functional diversity from the same RNA transcript. The percentage of modified RNA “transcript a” can be very low under one cellular condition (e.g. Cell state 1), but change to high (e.g. Cell state 2) under another cellular condition. By causing RNA structural changes and direct recruitment of modification reader proteins, the modified “transcript a” acquires a different fate, for example, from protein translation to increased RNA decay.
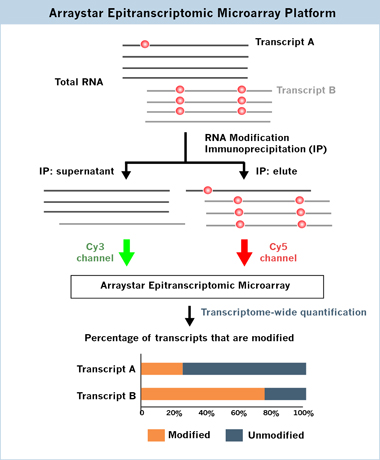
Fig 2. Arraystar Epitranscriptomic Microarray detects the immunoprecipitated modified RNA in Cy5 and supernatant unmodified RNA in Cy3 channels on the same array, such that the modified and unmodified percentages for each transcript can be measured. Alternatively spliced transcript isoforms are specifically and unambiguously detected by transcript-specific array probes.
Coverage of coding and noncoding epitranscriptomes
• Arraystar mRNA&lncRNA Epitranscriptomic Microarrays
For mRNA, lncRNA, and mid-sized noncoding RNA classes of pre-miRNA, pri-miRNA, snoRNA, and snRNA.
• Arraystar circRNA Epitranscriptomic Microarrays
For circular RNAs, at high confidence collection of observed expression in >= 2 experiments and >= 4 samples.
• High sensitivity and accuracy, even for RNAs difficult for MeRIP-seq (e.g. lncRNAs and circRNAs).
As a rule of thumb, the abundance of LncRNAs/circRNA junction sites in samples is too low to be accurate quantified by sequencing, and similarly for the abundance of IP-enriched LncRNAs/circRNA junctions.
What Are the Limitations of RNA-seq for LncRNA Profiling? >>
Why Use Microarray Over RNA-seq for Circular RNA Expression Profiling? >>
RNA modifications at transcript-specific level
Alternatively spliced transcript isoforms have tissue-specific expression and distinct biological functions. For example, TRIM9 short isoform (NM_052978), not the long isoform (NM_015163), selectively inhibits the production of pro-inflammatory cytokines in response to viral infection[5]. Changes in their percentages of modified transcript isoforms have been associated with biological functions and diseases.
Arraystar Epitranscriptomic Microarrays use specific exon or splice junction probes to reliably and accurately profile the RNA modification in each individual transcript isoform, defining a new level of details of epitranscriptomics (Fig. 3).
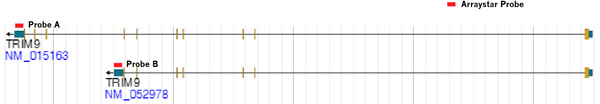
Fig 3. Arraystar Epitranscriptomic Microarray uses transcript-specific Probe A and Probe B to distinctly detect RNA modification in TRIM9 long (NM_015163) and short (NM_052978) transcript isoforms.
Low sample amount requirements
Current MeRIP-seq technique requires a massive amount of total RNA at >= 120 µg as the starting material, making it difficult to study on samples of limited availability. Arraystar Epitranscriptomic Microarray requires as little as is 3 µg total RNA, which is magnitudes lower than MeRIP-seq. The low sample amount requirement opens up opportunities for research projects where the samples are rare or of limited supply.
Arraystar Epitranscriptomic Microarray profiling is provided as a full service, from sample preparation, RNA immunoprecipitation/GST-YTH pull down, cRNA labeling, microarray experiment, to annotation and data analysis. The in-process QC steps are included to ensure data quality and success. Just send us your samples, and we’ll do the rest!
mRNA&lncRNA Epitranscriptomic Microarray Workflow
Take m6A modification as an example:
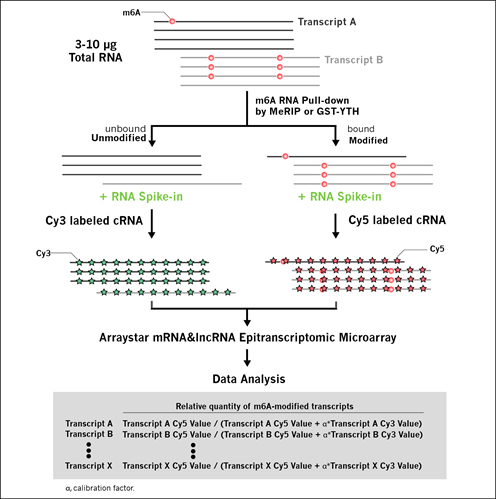
Fig 1. mRNA&lncRNA Epitranscriptomic Microarray Workflow.
• RNA samples provided by Customer (See Sample Submission for details)
• RNA QC
• m6A-enrichment by m6A-MeRIP or GST-YTH pulldown
• cRNA synthesis and two-color labeling (Cy5 for enriched-RNA and Cy3 for supernatant RNA)
• Array hybridization, washing, and scanning
• Data extraction, annotation, analysis and summarization
circRNA Epitranscriptomic Microarray Workflow
Take m6A modification as an example:
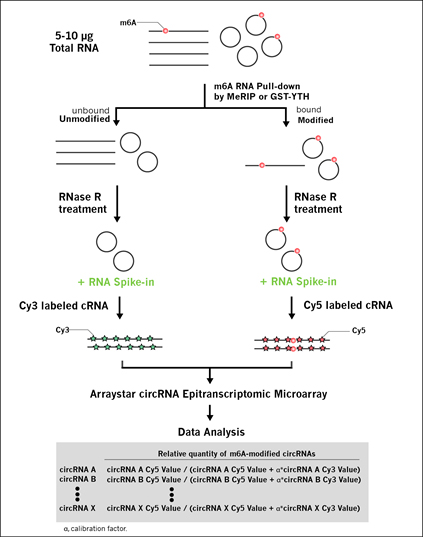
Fig 2. circRNA Epitranscriptomic Microarray Workflow.
• RNA samples provided by Customer (See Sample Submission for details)
• RNA QC
• m6A-enrichment by m6A-MeRIP or GST-YTH pulldown
• RNase R treatment to remove linear RNAs (e.g. rRNA, lncRNA, mRNA, etc)
• cRNA synthesis and labeling (Cy5 for enriched-RNA and Cy3 for supernatant RNA)
• Array hybridization, washing, and scanning
• Data extraction, annotation, analysis and summarization
Arraystar has the expertise and in-depth knowledge in microarray profiling, data analysis, and result interpretation. Rich and detailed epitranscriptomic bioinformatic analyses are provided in the data files and included in the service.
Differentially modified transcripts (mRNAs, lncRNAs, and mid-size ncRNAs)
![]()
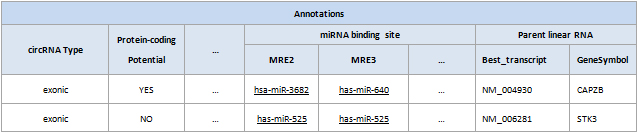
Differentially modified circRNAs
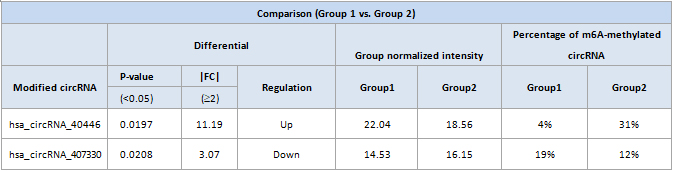
Hierarchical clustering heatmap of differentially modified RNAs

GO Enrichment Analysis of differentially modified mRNAs
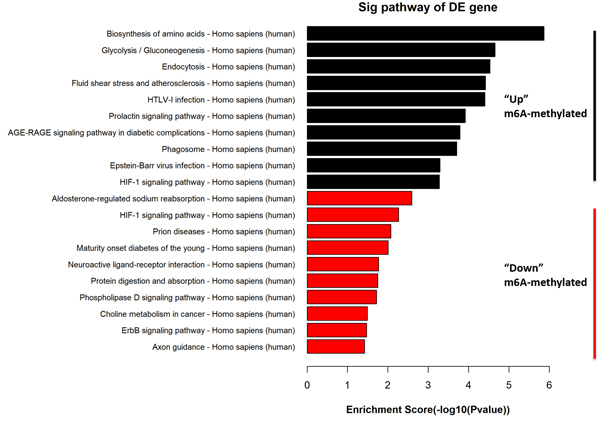
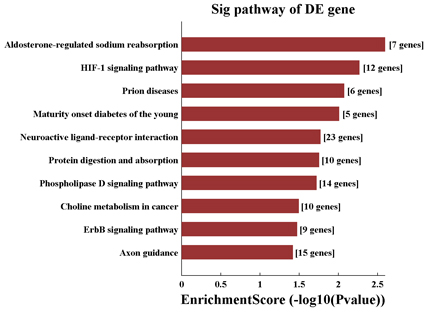
Pathway analysis of differentially modified mRNAs