
Cancer metabolism is one of the core hallmarks of cancer [1, 2]. Mainly driven by oncogenic signaling pathways and by amplified or alternatively spliced metabolic enzymes, the characteristic and profound metabolic alteration allows cancer cells to accommodate metabolic demands to sustain growth, proliferation, and survival in nutrient fluctuating environment [3, 4](Fig. 1). Enhanced uptake of glucose, a common feature of cancers, supports the production of intermediates for the synthesis of lipids, proteins and nucleic acids. In addition, increased glutamine uptake and glutaminolysis allow cancer cells to replenish intermediates in the tricarboxylic acid (TCA) cycle that are redirected into biosynthetic reactions. The increased biosynthetic activity also requires a corresponding increase in the production of NADPH as a reducing agent for anabolic reactions and to maintain cellular redox balance [5]. The epigenetic modifications during the process of cellular transformation and cancer progression are derived from metabolites such as acetyl-CoA for acetylation, NAD for deacetylation, SAM for methylation, α-ketoglutarate for demethylation, and UDP-GlcNAc for glycosylation [6].
Glucose uptake and glycolysis
Most cancers are programmed to increase glucose uptake and exhibit a high rate of glycolysis known as Warburg effect. Glycolysis provides the precursors for the biosynthesis of nucleotides, amino acids, and lipids. The very high rate of glycolysis allows the cells to maintain biosynthetic fluxes during rapid proliferation, while producing pyruvate in large amount. However, the high glycolytic flux may significantly exceed the maximum capacity of pyruvate dehydrogenase (PDH), an enzyme at the initiation of pyruvate oxidation in mitochondria [7]. To avoid pyruvate accumulation in cytoplasm, most of the pyruvate is converted to lactate by lactate dehydrogenase A (LDHA), a high-capacity system that removes pyruvate and regenerates NAD from NADH [8].
Lactate transport and pH regulation
Increased uptake of glucose and glycolysis create the problems of intracellular acidification and lactate accumulation. Maintaining an alkaline intracellular milieu is essential for continued cancer cell survival, whereas acidification of the extracellular microenvironment may facilitate cancer cell invasion and metastasis. Three main acid-regulatory systems regulate the pH in cancer cells: Na /H exchangers (NHE1), carbonic anhydrase 9 (CA9) and monocarboxylate transporters (MCTs). Lactate transport across the plasma membrane is facilitated by MCTs and is coupled to the symport of protons. Lactate produced by the hypoxic tumor cells can be taken up, oxidized, and used by cells within the oxygenated areas of solid tumours. Such lactate utilization increases the glucose availability to support the survival of cells within the hypoxic region of the tumour [5].
Gluconeogenesis and hexosamine metabolism
The glycolytic pathway can also flow in reverse to produce glucose from pyruvate, a process known as gluconeogenesis. As gluconeogenesis and glycolysis generate similar inter¬mediates, enhancement of either pathway could increase anabolic precursors for cell growth. Certain types of cancer cells utilize the gluconeogenic enzymes phosphoenolpyruvate carboxykinase (PEPCK) and phosphoenolpyruvate carboxykinase 2 (PCK2) to channel tricarboxylic acid (TCA) cycle-derived substrates into macromolecular synthesis [9-11].
In addition to being metabolized through glycolysis, fructose 6-phosphate can act as a nitrogen acceptor from glutamine and be converted to glucosamine 6-phosphate. Further metabolism through the hexosamine biosynthetic pathway results in UDP-N-acetylglucosamine (UDP-GlcNac). UDP-GlcNac can be used for N- and O-linked-glycosylation, including glycosylation of growth factor receptors in the endoplasmic reticulum. Such glycosylation promotes cell response to growth factors. O-GlcNAcylation is a central communicator of nutritional status, which controls key signaling and metabolic pathways that manifest cancer cell phenotypes [12]. Increased total O-GlcNAcylation is emerging as a general characteristic of cancer cells.
Pentose phosphate pathway and nucleotide synthesis
The pentose phosphate pathway (PPP) branched from glycolysis is required for the synthesis of ribonucleotides and a major source of NADPH [13]. The PPP plays a pivotal role in cancer cells to meet the anabolic demands of nucleotides and to combat oxidative stress.
The biosynthesis of purine nucleotide (ATP, GTP, dATP, and dGTP) builds on 5-phosphoribosyl-α-pyrophosphate (PRPP), an activated ribose product of PPP. The purine ring is successively constructed from glutamine, glycine, 10-formyl-THF, CO2, and aspartate. Of these precursors, glycine can be derived from the glycolytic intermediate 3-phosphoglycerate (3PG); 10-formyl-THF, a donor of one-carbon unit, acquires the carbon unit from either serine or glycine, both of which can be derived from 3PG.
In biosynthesis of pyrimidine nucleotides (CTP, UTP, dCTP, and dTTP), the pyrimidine ring is first assembled from bicarbonate, aspartic acid, and ammonia (usually hydrolyzed from glutamine), and then attached to the ribosyl group of PRPP. Of these precursors, aspartate can be acquired directly from the environment or generated from oxaloacetate in the TCA cycle. The carbons in oxaloacetate can be originated from glucose or glutamine. For thymidine, the one additional carbon is derived from 5,10-methyleneTHF [7].
Glycerol, fatty acid, and cholesterol synthesis (Lipid metabolism)
Lipid metabolism, in particular the synthesis of fatty acids (FAs), is an essential cellular process that produces metabolic intermediates for membrane biosynthesis, energy storage and the generation of signaling molecules. Deregulated FA synthesis contributes to cancer on multiple levels not only by making membrane lipids for active cell growth or by provisioning substrates for ATP synthesis, but also by signaling pathways as lipid signaling molecules (e.g. LPA, prostaglandins), second messengers (e.g. PIP3), or acyl modifiers on other signaling molecules (e.g. on WNTs) that regulate cell proliferation and survival [14]. Glycolysis produces much of the intermediates that convert to lipids. For example, dihydroxyacetone phosphate (DHAP) is the precursor to glycerol-3-phosphate, which is crucial for the biosynthesis of the phospholipids and triacylglycerols as the major structural lipids in cell membranes. 3-phosphoglycerate (3PG) is the precursor of sphingolipids, another major class of lipids in cells. Most of the acetyl-CoA for fatty acid synthesis is derived from conversion of glycolysis product pyruvate in mitochondria and carried across the membrane as citrate. Acetyl-CoA provides the carbon unit for synthesis of fatty acyl chain components of the various lipid classes and also for mevalonate (MVA), a precursor for cholesterol and related molecules [7]. The MVA pathway is essential to produce sterols and isoprenoids that are integral to tumour growth and progression. In recent years, many oncogenic signaling pathways have been shown to increase the activity and/or the expression of MVA pathway enzymes [15].
Serine, glycine and one-carbon metabolism
One-carbon metabolism involves interconnected folate cycle and methionine cycle. It transfers one-carbon group from amino acids, mostly serine and glycine, to support nucleotide synthesis, generation of methyl group donors for methylation reactions, antioxidant production and maintaining redox status (GSH synthesis and NADPH production), all of which are crucial to support cancer cell growth, tumour homeostasis and epigenetic modifications [16]. Serine and glycine can be de novo synthesized from the glycolytic intermediate 3-phosphoglycerate or imported as nutrients. Serine and glycine fuel the folate cycle by the pivotal serine to glycine conversion with serine hydroxymethyltransferase (SHMT) and by glycine cleavage with glycine dehydrogenase (GLDC). The folate cycle is then coupled to the methionine cycle with methyl-THF (mTHF) by donating a carbon group to homocysteine, converting it to methionine. S-adenosylmethionine (SAM), one of the methionine cycle intermediates, is the methyl group donor required in methylation modification of DNA, RNA, proteins, lipids as well as n-propylamine donor in polyamine synthesis [17]. Serine, glycine, and one-carbon metabolism are a central hub in cancer metabolsim. Many cancer cell types are highly dependent on serine synthesis or exogenous supply. Conversely, hyperactivation of serine/glycine biosynthesis drives oncogenesis. Antifolate chemotherapies in one-carbon metabolsim are an important strategy widely used in cancer treatment.
TCA cycle and cataplerosis
To synthesize lipids, proteins, and nucleic acids, proliferating cancer cells can use TCA cycle intermediates as the precursors diverted from the cycle (cataplerosis). Under normoxic conditions, glucose-derived acetyl-CoA and glutamine-derived oxaloacetate condense to form citrate by citrate synthase (CS) to enter the TCA cycle. However, oxidative mitochondrial metabolism can be truncated or impaired in cancer cells as a result of mutations in components of the TCA cycle or electron transport chain. Moreover, tumour hypoxia inhibits the entry of pyruvate into the TCA cycle and pre¬vents the synthesis of citrate through this route. Under these condi¬tions, reductive carboxylation of glutamine-derived α-ketoglutarate by mitochondrial NADPH-dependent isocitrate dehydrogenase (IDH2) is used to generate citrate for lipid synthesis [5], where citrate is exported to the cytosol and converted to acetyl-CoA for fatty acid synthesis.
Glutamine uptake, glutaminolysis and anaplerosis
Many cancer cells have dependence on glutamine and high glutaminolytic flux rates referred to as glutamine addiction. Cancer cells often rely on aerobic glycolysis; however the metabolites upstream of pyruvate are diverted and pyruvate is converted to lactic acid. The decrease of pyruvate entering the TCA cycle can limit the metabolic intermediate supply for the synthesis of fatty acids, proteins, and nucleotides. In such a case, glutamine anaplerosis is key to refill the TCA cycle with the concurrent production of reduced NADPH. Glutamine transported into the cells is metabolized by glutaminolysis to α-ketoglutarate (α-KG) by glutamate dehydrogenase (GLUD) or by aminotransferase, α-KG is converted to citrate with the consumption of NADPH, a reductive carboxylation process in the reverse TCA cycle. Citrate is converted by ATP-citrate lyase (ACL) to acetyl-CoA. Thus, by contributing to anaplerotic flux, glutamine provides carbons for TCA cycle intermediates that serve as precursors of acetyl-CoA for fatty acids, aspartate from oxaloacetate for nucleotides synthesis, and the synthesis of many nonessential amino acids. Furthermore, glutamine is a nitrogen source for nucleotides, amino acids, and hexosamines [7].
Biosynthesis of nonessential amino acids (NEAA) and mTORC1 signaling
Most nonessential amino acids are produced through transamination reactions. Glycolytic intermediates 3-phosphoglycerate, pyruvate, and phosphoenolpyruvate are carbon skeleton precursors for the biosynthesis of serine, glycine and cysteine, alanine, and tyrosine, while mitochondrial intermediates α-KG and oxaloacetate for glutamate, glutamine, proline, arginine, aspartate, and asparagine. Most cancers depend on these syntheses rather than exogenous supplies.
Amino acid availability provides the dominant signal input for the cell growth signal transduction machinery. The mammalian target of rapamycin protein complex 1 (mTORC1) is a serine/threonine kinase that coordinates cell growth with the availability of amino acids (especially for arginine, glutamine, and leucine) and the growth signals [7]. The amino acid-sensing complex that contains the Rag GTPases, Regulator, and the v-ATPase on the lysosome is at the core of mTORC1 activation. mTORC1 is highly activated in proliferating cancer cells, which activates protein synthesis , mitochondrial biogenesis and de novo lipogenesis.
Bioenergetics and redox balance
In cancer cells, ATP generated from aerobic glycolysis gives tumors a growth advantage under hypoxia or fluctuating oxygen availability. Most cancer cells retain functional mitochondria and in fact mitochondria still generate the majority of ATP in many cancer cells [1]. However by oncogenic reprogramming of the metabolism, ATP production by oxidative phosphorylation becomes only secondary to the synthesis of anabolic precursors.
Unlike ATP, cytosolic NADPH can be limiting for cell proliferation. NADPH is the essential reducing equivalents for fatty acid and cholesterol biosynthesis, as well as for modulating oxidative stress. In proliferating cells, the pentose phosphate pathway (PPP) is the largest producer of cytosolic NADPH. In many cancer cells under oxidative stress, pyruvate kinase isoform M2 (PKM2) is often expressed and diverts glycolysis to pentose phosphate pathway to produce NADPH. Surprisingly, serine-driven one-carbon metabolism involving folate cycle is a nearly comparable major source of NADPH generation [18]. Additionally, cytoplasmic NADPH can be produced by oxidative decarboxylation of malate to pyruvate catalyzed by malic enzyme 1 (ME1) or by conversion of citrate to α-ketoglutarate by IDH1. In mitochondria where the NADH:NAD ratio is high particularly under hypoxia, the TCA cycle electron carrier output NADH can be converted to NADPH by mitochondrial nicotinamide nucleoside transhydrogenase (NNT). Additional NADH can also be imported as the glycolytic product from cytosol by the aspartate–glutamate and malate–α-ketoglutarate shuttle system, which at the same time restores cytoplasmic pools of NAD to support the high glycolytic flux of cancer cells.
Oncogenic signaling drives cancer metabolic reprogramming
Within the hierarchy of pathways altered in cancer, glucose and glutamine metabolism are consistently reprogrammed by mutations of oncogenes and tumor suppressors. For example, oncogenic activation of AKT by phosphatidylinositol-3-OH kinase (PI3K) promotes glucose uptake, hexokinase activity and mitochondrial localization, and glyco¬lytic flux [19]. The mammalian target of rapamycin complex 1 (mTORC1) and hypoxia-inducible factor (HIF) up-regulate the expression and activity of glycolytic enzymes. In MYC-transformed cells, oncogenic levels of MYC increases glutaminolysis through a transcriptional program that results in glutamine addiction. MYC also promotes the alternative splicing of the pyruvate kinase embryonic isoform PKM2, which is highly or exclusively expressed in cancer cells. In response to tyrosine kinase signaling, PKM2 isoform has the distinctive property of switching from tetrameric to dimeric form at lower activity, consequently allowing accumulation of glycolytic intermediates for biosynthesis pathways [5].
Tumour suppressor pathways also affect metabolism. For example, p53 inhibits aerobic glycolysis, upregulates mitochondrial OXPHOS, and supresses fatty acid synthesis, which directly oppose many effects of the metabolic alterations crucial for malignant transformation and cancer progression [20]. p53 regulates glycolysis by inducing the expression of the TP53-induced glycolysis and apoptosis regulator (TIGAR), an enzyme with homology to fructose-2,6-bisphosphatase, that functionally inhibits glycolytic activity [5]. p53 also maintains mitochondrial activity through the expression of cytochrome c oxidase 2, the loss of which recapitulates the Warburg effect [5].
Metabolic enzymes acting as oncoproteins and tumor suppressors
In addition to the growth promoting metabolic alterations by oncogene activation, metabolic enzymes themselves can actively drive the transformation process. A large number of metabolic enzymes are amplified, alternatively spliced, or mutated in the pathogenesis of cancer. The gene copy numbers of hexokinase II (HK2), fatty acid synthase (FASN), and phosphoglycerate dehydrogenase (PHGDH) are amplified in cancer cells, contributing to heightened glycolysis, fatty acid synthesis and serine synthesis. Pyruvate kinase isoform M2 (PKM2) is highly expressed in rapidly proliferating tissues, and many cancer cells exclusively express this isoform [21]. As mentioned previously, PKM2 can switch from a tetrameric to a dimeric form with lower activity, allowing the accumulation of glycolytic intermediates for biosynthetic uses [22]. Other metabolic genes, including PFKFB3 [23] and GLS [24], also have alternatively spliced isoforms for cancer proliferation. Metabolic enzymes in the TCA cycle, such as fumarate hydratase (FH) and succinate dehydrogenase (SDH), can act as tumour suppressors. Loss-of-function mutations of FH and SDH can lead to accumulation of fumarate and succinate, which impair prolyl hydroxylases (PHDs), a α-ketoglutarate-dependent enzyme that modifies HIF-αstability. Fumarate can also modify the cysteine residues of the Keap1-Nrf2 complex, the major regulator of cytoprotective responses to oxidative and xenobiotics stress, causing prolonged activation of Nrf2 having dual roles in cancer progression. Isocitrate dehydrogenases IDH1 and IDH2 catalyze the oxidative decarboxylation of isocitrate to α-KG. Mutant forms of IDH1 or IDH2 frequently found in glioma and acute myeloid leukemia nearly completely abolish the normal activity to produce α-KG while at the same time gain the new function of reducing α-KG to 2-hydroxyglutarate (2-HG) [5]. Oncometabolite 2-HG generated by this neomorphic enzymatic activity competitively inhibits α-KG-dependent prolyl hydroxylases (PHDs), an enzyme that can hydroxyl modify and target HIF-1 for degradation. Thus IDH1 and IDH2 oncogenic mutations have the outcome of ultimately raising HIF-1 levels in cancer cells. Additionally, 2-HG is also a competitive inhibitor of histone demethylases and the TET family of 5-methlycytosine (5mC) hydroxylases, causing genome-wide histone and DNA methylation alterations and epigenetic dysregulation in the cancers [25].
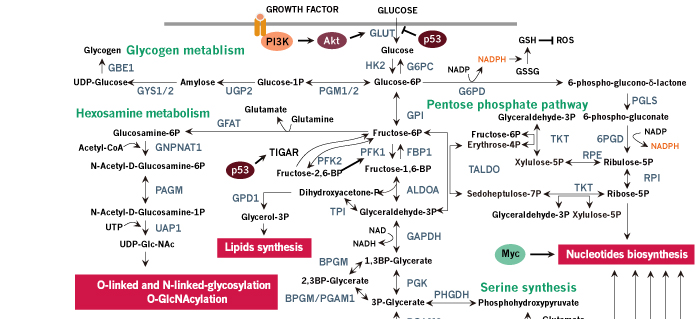
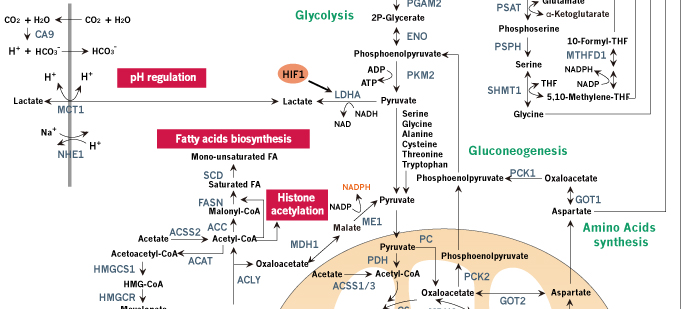
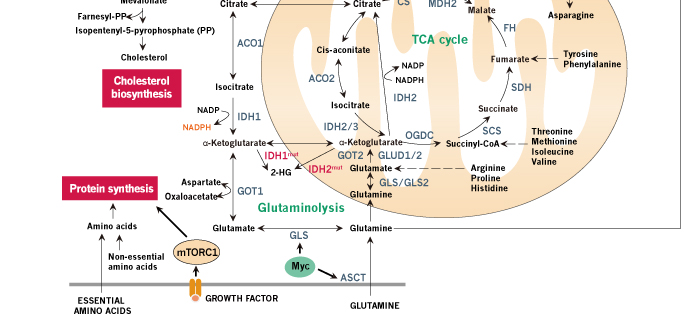
Figure 1. Schematic review of cancer metabolism. Glucose, glutamine and other nutrients metabolism are consistently reprogrammed by mutations of oncogenes and tumor suppressors to support growth and proliferation of cancer cells. Oncogenic activation of AKT by PI3K results in increased glucose uptake and enhanced glycolytic flux, while the glycolytic intermediates are shunted into biosynthetic pathways and other subsidiary pathways. Biosynthetic pathways including pentose phosphate pathway, serine biosynthetic pathway and glycerol biosynthetic pathway provide anabolic precursors for nucleotides, amino acids and lipids, while subsidiary pathways like hexosamine pathway and glycogen pathway support tumor progression. In cancer cells, mitochondria function as biosynthetic organelles and TCA cycle are redirected to anabolic reactions, generating important building blocks such as aspartate and acetyl-CoA. Aspartate can be used for protein synthesis and is essential for nucleotide synthesis, and acetyl-CoA provides the carbon for synthesis of fatty acids and cholesterol. The continuous efflux of TCA intermediates can be replenished by glucose or glutamine. Oncogenic levels of Myc promote glutamine uptake and glutamine metabolism, thereby refilling the TCA cycle and supplying nitrogen for cancer cells. In addition to de novo amino acid synthesis, cancer cells increase the uptake of other amino acids such as branched-chain amino-acids (BCAAs). The amino acids and oncogenic growth-factor signaling together activate mTORC1 in the membrane of lysosome, promoting protein synthesis and de novo lipogenesis. Importantly, when glucose or other nutrients are scarce, cancer cells use amino acids, especially BCAAs, to refill the TCA cycle and hijack the gluconeogenic pathway to provide glycolytic intermediates. Tumor suppressor p53 opposes the metabolic alteration in cancer by inhibiting glucose transport and inducing the expression of TIGAR. NADPH production is increased in cancer cells for anabolic reactions and redox balance maintenance. Several pH-regulatory systems maintain cellular pH homeostasis. Metabolites such as acetyl-CoA and UDP-GlcNAc are the chemical group donors of oncogenic modifications of protein, DNA, and chromatin.
Related Product:
NuRNA™ Central Metabolism PCR Array (H)
Abbreviations:
ACAT, Acetyl-CoA acetyltransferase; ACC, Acetyl-CoA carboxylase ; ACLY, ATP-citrate synthase; ACO, Aconitase; ACSS, Acetyl-coenzyme A synthetase; ALDOA, Aldolase A; ASCT, Alanine/serine /cysteine/threonine transporter 1; BPGM, Bisphosphoglycerate mutase; CA9, Carbonic anhydrase 9; CS, Citrate synthase; ENO, Enolase; FASN, Fatty acid synthase; FBP1, Fructose-1,6-bisphosphatase 1; FH, Fumarate hydratase; G6PC, Glucose-6-phosphatase; G6PD, Glucose 6-phosphate dehydrogenase; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; GBE, Glycogen-branching enzyme; GFAT, Glutamine:fructose-6-phosphate amidotransferase ; GLS, Glutaminase; GPD: Glucose-6-phosphate isomerase; GLUD, Glutamate dehydrogenase; GLUT, Glucose transporter; GNPNAT1, Glucosamine 6-phosphate N-acetyltransferase; GOT, Glutamate oxaloacetate transaminase; GPI, Glucose-6-phosphate isomerase; GSH, Reduced glutathione monomer; GSSG, Oxidized glutathione dimer; GYS, Glycogen [starch] synthase; HIF1, Hypoxia inducible factor 1; HK, Hexokinase; HMGCR, 3-Hydroxy-3- methylglutaryl-coenzyme A reductase; HMGCS, Hydroxymethylglutaryl-CoA synthase; IDH, Isocitrate dehydrogenase; LDHA, L-lactate dehydrogenase A; MCT1, Monocarboxylate transporter 1; MDH, Malate dehydrogenase; ME1, Malic enzyme 1; MTHFD, Methylenetetrahydrofolate dehydrogenase; mTORC1, Mechanistic target of rapamycin complex 1; NHE1, Na( )/H( ) exchanger 1; OGDC, 2-Oxoglutarate dehydrogenase complex; p53, Tumor suppressor p53; PAGM, Phosphoacetylglucosamine mutase; PC, Pyruvate carboxylase; PCK, Phosphoenolpyruvate carboxykinase; PDH, Pyruvate dehydrogenase; PFK, Phosphofructokinase; PGAM, Phosphoglycerate mutase ; PGK, Phosphoglycerate kinase; PGLS, 6-Phosphogluconolactonase; PGM, Phosphoacetylglucosamine mutase; PHGDH, D-3-phosphoglycerate dehydrogenase; PI3K, Phosphoinositide-3- kinase; PKM2, Pyruvate kinase isozyme M2 ; PSAT, Phosphoserine aminotransferase; PSPH, Phosphoserine phosphatase; ROS, Reactive oxygen species, RPE, Ribulose 5-phosphate 3-epimerase; RPI, Ribose-5-phosphate isomerase; SCD, Stearoyl-CoA desaturase; SCS, Succinyl-CoA synthetase; SDH, Succinate dehydrogenase; SHMT, Serine hydroxyl-methyltransferase; TALDO, Transaldolase; TIGAR, TP53-induced glycolysis and apoptosis regulator; TKT, Transketolase; TPI, Triosephosphate isomerase; UAP1, UDP-N-acetylhexosamine pyrophosphorylase; UGP, UDP-glucose pyrophosphorylase; 2-HG, 2-Hydroxyglutarate; 6PGD, 6-Phosphogluconic dehydrogenase.
References
1. Ward, P. S. and Thompson, C. B. (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell, 21(3):297-308 [PMID: 22439925]
2. Pavlova, N. N. and Thompson, C. B. (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab, 23(1):27-47 [PMID: 26771115]
3. Boroughs, L. K. and DeBerardinis, R. J. (2015) Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol, 17(4):351-9 [PMID: 25774832]
4. Sullivan, L. B., et al. (2016) Altered metabolite levels in cancer: implications for tumour biology and cancer therapy. Nat Rev Cancer, 16(11):680-693 [PMID: 27658530]
5. Schulze, A. and Harris, A. L. (2012) How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature, 491(7424):364-73 [PMID: 23151579]
6. Kinnaird, A., et al. (2016) Metabolic control of epigenetics in cancer. Nat Rev Cancer, 16(11):694-707 [PMID: 27634449]
7. Lunt, S. Y. and Vander Heiden, M. G. (2011) Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol, 27(441-64 [PMID: 21985671]
8. DeBerardinis, R. J., et al. (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab, 7(1):11-20 [PMID: 18177721]
9. Montal, E. D., et al. (2015) PEPCK Coordinates the Regulation of Central Carbon Metabolism to Promote Cancer Cell Growth. Mol Cell, 60(4):571-83 [PMID: 26481663]
10. Balsa-Martinez, E. and Puigserver, P. (2015) Cancer Cells Hijack Gluconeogenic Enzymes to Fuel Cell Growth. Mol Cell, 60(4):509-11 [PMID: 26590709]
11. Vincent, E. E., et al. (2015) Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol Cell, 60(2):195-207 [PMID: 26474064]
12. Ward, P. S. and Thompson, C. B. (2012) Signaling in control of cell growth and metabolism. Cold Spring Harb Perspect Biol, 4(7):a006783 [PMID: 22687276]
13. Patra, K. C. and Hay, N. (2014) The pentose phosphate pathway and cancer. Trends Biochem Sci, 39(8):347-54 [PMID: 25037503]
14. Rohrig, F. and Schulze, A. (2016) The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer, 16(11):732-749 [PMID: 27658529]
15. Mullen, P. J., et al. (2016) The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer, 16(11):718-731 [PMID: 27562463]
16. Locasale, J. W. (2013) Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer, 13(8):572-83 [PMID: 23822983]
17. Amelio, I., et al. (2014) Serine and glycine metabolism in cancer. Trends Biochem Sci, 39(4):191-8 [PMID: 24657017]
18. Fan, J., et al. (2014) Quantitative flux analysis reveals folate-dependent NADPH production. Nature, 510(7504):298-302 [PMID: 24805240]
19. Elstrom, R. L., et al. (2004) Akt stimulates aerobic glycolysis in cancer cells. Cancer Res, 64(11):3892-9 [PMID: 15172999]
20. Berkers, C. R., et al. (2013) Metabolic regulation by p53 family members. Cell Metab, 18(5):617-33 [PMID: 23954639]
21. Mazurek, S. (2011) Pyruvate kinase type M2: a key regulator of the metabolic budget system in tumor cells. Int J Biochem Cell Biol, 43(7):969-80 [PMID: 20156581]
22. Christofk, H. R., et al. (2008) Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature, 452(7184):181-6 [PMID: 18337815]
23. Zscharnack, K., et al. (2009) The PFKFB3 splice variant UBI2K4 is downregulated in high-grade astrocytomas and impedes the growth of U87 glioblastoma cells. Neuropathol Appl Neurobiol, 35(6):566-78 [PMID: 19490427]
24. Cassago, A., et al. (2012) Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A, 109(4):1092-7 [PMID: 22228304]
25. Wallace, D. C. (2012) Mitochondria and cancer. Nat Rev Cancer, 12(10):685-98 [PMID: 23001348]