
The lncRNA transcription is tightly regulated in a cell type-, tissue-, or disease-specific manner, much more so than the transcription of protein coding genes. Understanding what is behind the lncRNA expression dysregulation and the upstream regulatory mechanisms is a key part of lncRNA research. Epigenetic study of transcriptional regulation on lncRNAs is an important avenue from the genomic perspective.
DNA methylation or hydroxymethylation in the promoter regions critical for transcription have the regulatory effects on gene expression. For example, the methyl group modification in the LncRNA-MEG3 promoter region is aberrantly changed in liver cancers and schizophrenia patients [1]. As biomarkers, the lncRNA promoter methylation signature has shown classifying power between tumor and non-tumor cells higher than mRNA promoters in esophageal cancers [2]. Dysregulation of lncRNA promoter methylation is common, present in up to 57% breast cancer specimens [3]. Not surprisingly, the levels of lncRNA promoter methylation and the lncRNA gene expression are significantly correlated.
Arraystar (h)MeDIP-seq service with lncRNA promoter analysis offers added benefits to obtain in depth data analyses and annotations of 5mC or 5hmC modifications on both mRNA and lncRNA gene promoters(Figure.1). Moreover, the analysis can be combined and integrated seamlessly with Arraystar LncRNA Array gene expression profiling of both lncRNA and mRNA, to systematically investigate the epigenetic regulation and gene expression in the biological system or disease.
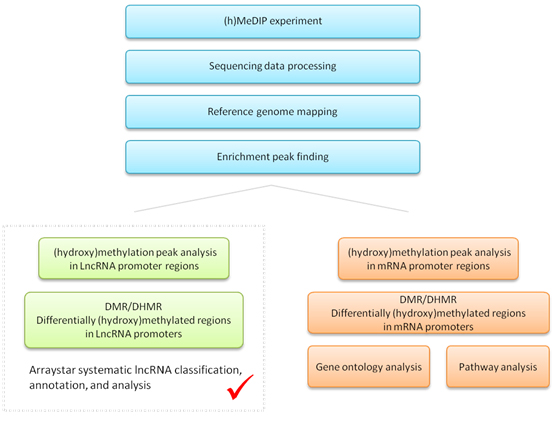
Figure 1. The workflow of Arraystar MeDIP/hMeDIP-seq, which distinguishes itself with lncRNA promoter analysis along with mRNA promoters.
References
1.Thomson, J.P. et al.(2013) Nucleic Acids Res 41(11):5639-54 [PMID:23598998]
2.Wu, W. et al.(2013) Gastroenterology 144(5):956-966.e4 [PMID:23333711]
3.Li, Y. et al.(2015) Scientific Reports 5;5:8790 [PMID:25739977]
Differential methylation or hydroxymethylation in lncRNA promoter regions between samples or groups
The differential (hydroxy)methylation in the lncRNA promoter regions ( /- 2000 bp of TSS) is annotated with the reference genome (Fig. 1).
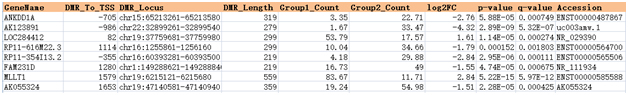
Figure 1. Differentially methylated regions in the lncRNA promoter regions are tabulated with the fold changes , p- and q-values for statistical significance, and genomic annotation.
Combined analysis of (h)MeDIP-seq of lncRNA promoters with lncRNA expression
The epigenetic modification data from (h)MeDIP-seq and the gene expression profiles are combined for integrative analysis. The 5mC, 5hmC or 6mA differentially modified genes and the differentially expressed genes are hierarchically clustered on the heatmaps, to allow correlation of the changes (Fig. 2).
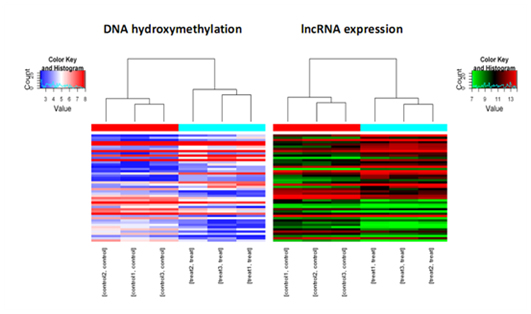
Figure 2. A combined analysis of hMeDIP-seq and LncRNA expression array profiling, as shown in the clustering heatmaps, to reveal the relationship of hydroxymethylation of lncRNA promoters with the lncRNA gene expression.
Visualization of LncRNA promoter (hydroxy)methylation
The (h)MeDIP-seq data are formatted as compatible WIG files for uploading to genome browsers such as UCSC Genome Browser. The (h)MeDIP signals can be visualized along with genomic features and a wealth of other gene expression, gene regulation, phenotypic tracks from many other sources.
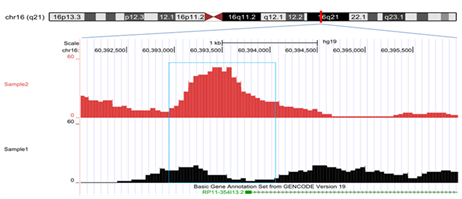
Figure 3. Genome browser visualization of hydroxymethylation peaks from hMeDIP-seq data.