
Genome-wide peak or binding site detection with around 10 million uniquely aligned reads
Our ChIP-seq generates around 10 million uniquely aligned reads per ChIP-DNA sample. In order to identify the protein binding sites from ChIP-seq data, we use Model-based Analysis of ChIP-Seq (MACS) to predict the protein-DNA interaction sites. This is useful for ChIP-seq peak detection without the use of controls. MACS provides detailed information for each peak, such as genome coordinates, p-value and summits (peak center). The discrete regions of 150 bp around each ChIP-seq peak are defined as protein binding sites.
Annotation and distribution of protein binding sites in relation to gene annotation
By mapping the binding sites in relation to Refseq genes, we can provide the annotation and distribution of TF binding sites across five different categories of genomic elements including the promoter, exon, intron, upstream and intergenic regions. TF binding profiles around TSS (Transcription Start Sites) are also provided.
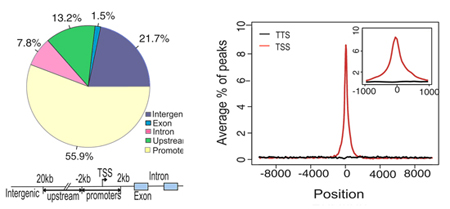
ChIP-peak distribution: Left: ChIP-peak distribution across five different genomic regions. The definition of each genomic region is described below. Core promoters are within -2kb to 2kb from the TSS. Upstream is from 2 to 20 kb from the TSS, and intergenic is a region not included as a promoter, upstream, intron or exon. Right: Shows the distribution of TF binding profiles within -10kb to 10 kb from the TSS (Inset shows a close up of a 1kb region centered on the TSS)
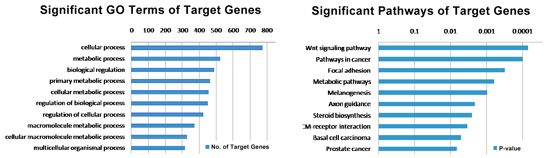
Putative TF target detection and their functional analysis
In order to investigate the function of TF suggested by its genome-wide binding site profile, we first identify target genes that have binding sites on their promoters (-2kb to 2kb from the TSSs). We then analyze the functional categories of these TF putative target genes by using GO enrichment analysis or pathway analysis.
Motif analysis of TF binding sites
In order to discover TF motifs, we perform a de novo motif search using the discovery rank of the imbalanced motifs (DRIM). We first classify TF binding sites into three different groups (strong, moderate and weak) based on their binding strength. Next, we extract the top 500 sites from each group and then perform DRIM on each set separately. This analysis is not included in the standard analysis package, and may involve additional charges.

Examples of DRIM identified novel motif. ChIP-sequencing is able to identify the TFBS (transcription factor binding site) motif patterns based upon the sequencing data. The motif logo above shows the typical results for a binding motif. The nucleotides are sorted by their frequencies from top to bottom, and the height of each nucleotide is proportional to its frequency. The top nucleotide is the most enriched at these positions.
High-Resolution visualization of binding profiles within regions of interest
Using UCSC genome browser, one can visualize our ChIP-seq data together with gene structure, conservation score, histone modification and other transcription factor binding signals.

ChIP-seq signal visualization. ChIP-Seq signals of Dpy-30 and H3K4me3 are visualized together in the genome browser. Representation of gene loci includes the Pcdhg gene cluster. The visualization of ChIP-Seq binding clearly shows their strong genome-wide overlap in both peak distribution and relative heights. (Jiang, H., et al., Cell, 2011. 144(4): p. 513-525. Dpy-30 library construction, sequencing and basic data analyses were performed at Arraystar Inc.)
References
1. Johnson, D.S., et al., Genome-wide mapping of in vivo protein-DNA interactions. Science, 2007. 316(5830): p. 1497-502.
2. Robertson, G., et al., Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods, 2007. 4(8): p. 651-7.
3. Barski, A., et al., High-resolution profiling of histone methylations in the human genome. Cell, 2007. 129(4): p. 823-37.
4. Mikkelsen, T.S., et al., Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 2007. 448(7153): p. 553-60.
Your Samples:
Chromatin immunoprecipitated DNA enriched by specific antibodies
Please refer to Sample Submission for details in how to get your project started.
Arraystar ChIP-seq:
2. Sequencing library prep and library QC
3. Sequencing by Illumina platform
4. Data Extraction, Analysis and Summarization
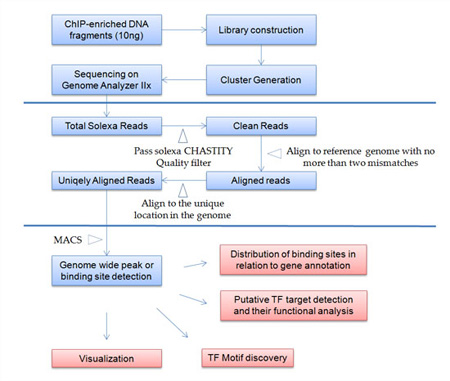
Figure. Flowchart of ChIP-seq Service at Arraystar