
The high throughput sequencing technique provides high sensitivity and specificity to analyze the abundance of microRNA sequence in a sample as well as to discover novel microRNA species. Arraystar offers Integrated microRNA Sequencing Service from sequencing library preparation to comprehensive data anlaysis. Our data analysis process generally consists of the following steps, raw data processing, usable reads filtering (including 3′ adapter trimming and normalization), and bioinformatics analysis( fig. 1).
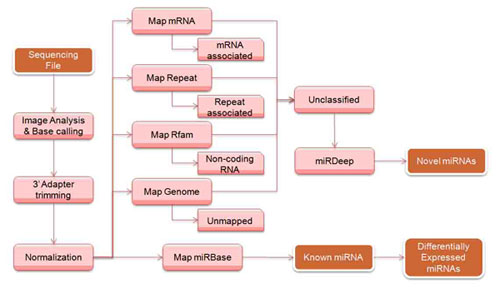
Fig. 1 Work flow of profiling microRNA from high throughput sequencing data.
1. Raw Data Processing.
The Genome Analyzer generates all the sequencing raw data in TIFF format. To extract the sequence information from the original image files, image analysis and base calling are performed using solexa pipeline V1.6 (OFF-LINE BASE CALLER V1.6 SOFTWARE). The short reads passed solexa CHASTITY filtering are retained for further processing.

2. 3′ adapter Trimming.
In the first step, the reads which pass the Solexa CHASTITY filtering are then passed through an adaptor filter that searches for reads whose 3′-ends align to the 3′-adaptor. The adaptor sequences are trimmed and then the adaptor-trimmed reads which have passed both filters are formatted into a non-redundant fasta file where the copy number and sequence is recorded for each unique tag. All sequences that fail to meet the quality cutoff, read length < 16nt and copy number < 2 are discarded as unusable reads.
After 3′ adapter trimming, a bar graph of sequence reads length distribution(Fig. 2) is used to show the overview of sequencing data.
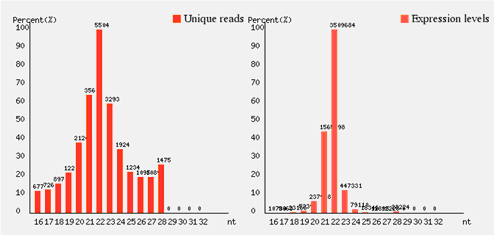
Fig. 2 microRNA sequencing data from a human sample. The length distribution of the reads is centred on 22 nt, this illustrates microRNA sequencing is reliable. The sequencing data is shown in unique reads and expression levels. Unique reads: sequence reads of a particular type with non-redundancy. Expression levels: All the reads from sequencing data, reflects relative abundance.
3. Normalization.
For each microRNA sequence-based profile, the copy number of small RNA sequence reads can be used to estimate expression level of each microRNA. Given a set of sequence-based profiles, a normalization step is needed for profile-to-profile comparisons.
Generally, the total number of valid sequence reads for each profile (be they from microRNA or other RNA species) is used as the scaling factor for normalization [(total sequence reads, given)/(total sequence reads, reference)], as this number would given an indication of the total amount of RNA in the given sample. One profile is used as the reference, and the values of each of the other profiles are divided by the scaling factor. The assumption made here is that the total small RNA population remains fairly constant between samples (without necessarily trying to make assumptions on the microRNA sub-population).

4. Bioinformatics Analysis.
After normalization, the basic bioinformatics analysis of microRNA sequencing data is performed, this generally contains two parts:
4.1 Identification of differentially expressed known miRNAs
4.2 Discovery of novel miRNAs
4.1 Identification of differentially expressed known miRNAs
A. Aligning to the Sanger miRBase: The usable reads are firstly aligned to the latest sanger microRNA database to identify all the known microRNA species in sequencing samples and proceed for further expression analysis; After mapping to the miRBase, a statistic table (Table. 1) is given to show the overview of sequencing data.
C1 | C3 | H1 | H3 | |
pass solexa CHASTITY quality filter | 6,634,171 | 6,803,894 | 4,812,612 | 6,365,678 |
3′ Adaptor trimming | 6,007,488 | 6,628,544 | 3,425,020 | 5,810,450 |
alignment miRBase 14 | 5,943,027 | 6,594,948 | 3,367,079 | 5,778,703 |
Table 1 A statistic table of microRNA sequencing data from 4 human samples. C1, C3, H1 and H3. The number in the table represent the reads number after each filtering step, including solexa CHASTITY quality filter, 3′ Adapter trimming and miRBase 14 alignment.
B. Detailed annotation of known miRNAs: After mapping to the miRBase, visual sequence alignments matched to each specific pre-miRNA in miRBase and detailed annotation(Fig.3) are further described. These information mainly includes:
* Structure and sequence information of pre-microRNA in miRBase
* Tag_counts of each isomiR: Copy number of each isomiR mapped to a specific pre-miRNA in miRBase
* Overlapped_Tag count: total number of all sequence tags overlaped with a specific microRNA form (3p or 5p).
* Most abundant_Tag count: the most abundant tag ID mapped to a specific microRNA.
* MaxCountIsoform: Sequence of the most abundant isomiR of a given microRNA

Fig. 3 the sequence aligned to miRBase are organized by mapping to the microRNA precursor sequence (hsa-let-7d here). As shown in the figure, the output data provide the information of each pre-miRNA including ID, genomic location, folding energy, secondary structure, and each sequenced reads mapped to the pre-miRNA. The mature miRNA form and star form in miRBase reference are indicated by ” ” and “*”. The normalized expression level from each sample estimated by the number of read counts are also given at the right panel.
miRNAs frequently exhibited variation from their “reference” sequences, producing multiple mature variants that we refer to as isomiRs. It appears that much of the isomiR variability can be explained by variability in either Dicer1 or Drosha cleavage positions within the pre-miRNA hairpin.
Because choosing a different isomiR sequence for measuring miRNA expression level can affect the ability to detect differential miRNA expression, we provide 3 different counting methods for miRNA expression level, using the copy number of the most abundant isomiR, all overlapped reads or the mature form in database. However, it is reported that the read count for the most abundant isomiR, rather than the miRBase reference sequence, provides the most robust approach for comparing miRNA expression between libraries.

C. Differential Expression Analysis
The absolute number of sequence reads for a particular microRNA represents a measure of its relative abundance. When comparing two groups of profile differences (such as disease versus control), microRNA levels may be determined by a two-sided t-test. The ‘fold change’ in microRNA expression between groups (the ratio of the group averages) may also be computed. When comparing sample groups, one may wish to consider the absolute microRNA levels as well as the relative levels; a change from 1000 to 2000 units is arguably more biologically relevant than a change from 5 to 10 units. In order to filter out from the analysis those microRNAs expressed at very low levels, one option is to add a given low number (e.g. 40 units) to each microRNA value [in the preceding example, (2000 40)/(1000 40)=1.96~2, while (10 40)/(5 40)=1.1]; alternatively, one could require a microRNA to be present at a minimum number of units in a given number of samples.

Examples
microRNA sequencing was performed on 2 human tissue samples C1 and C3, dataset of known microRNAs was obtained by aligning to Sanger miRBase 14. After normalization by the total number of each dataset, ratios of miRNA copy numbers between samples were obtained, as shown in the example table (Table.2) below.
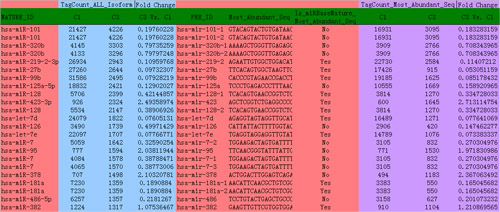
Table 2. Expression Level Analysis between microRNA sequencing samples. This table is a screen shot of the XLS file from mature miRNA-centered annotation that was process for better visualization. For each microRNA sequence-based profile, the absolute number of sequence reads for a particular miRNA represents a measure of its relative abundance. Fold change was used to profile differences miRNA levels between samples. MATURE_ID: ID from Sanger miRBase; TagCount_ALL_Isoform: total number of all sequence tags mapped to MATURE_ID of current line; PRE_ID: pre-microRNA ID from miRBase; Is_miRBaseMature_Most_Abundant_Seq: this shows whether the most abundant tag sequence information the same as the mature microRNA sequence in the miRBase; TagCount_Most_Abundant_Seq: the total number of the most abundant tags mapped to current entry; Fold Change: Fold change of miRNA expression between groups, calculated separately using both TagCount_ALL_Isoform and TagCount_Most_Abundant_Seq. In order to filter out the microRNA with low expression level, 10 is added to each tag count before calculating the fold change.
4.2 Novel miRNA Prediction.
The mechanism of novel microRNA discovery
The most intriguing feature of deep sequencing technology is to discover potential novel microRNAs from huge sequencing data. This can be achieved by miRDeep algorithm[1]. By using a simple model for miRNA precursor processing by Dicer, miRDeep is capable of both recovering the majority of known miRNAs present in heterogeneous deep-sequencing samples and reporting novel miRNAs with high confidence(Fig 4).
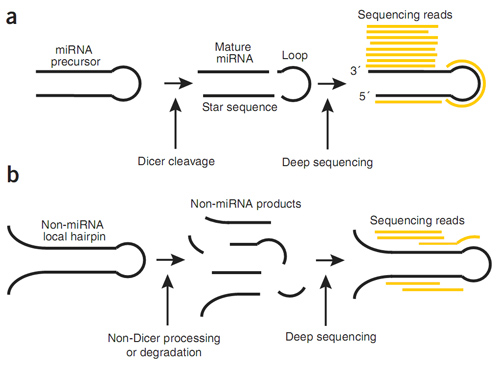
Fig. 4. Analyzing the compatibility of sequenced RNAs with miRNA biogenesis. (a) Each of the RNA products generated after a stable miRNA precursor is cleaved by Dicer the mature miRNA sequence, the star sequence and the loop as a certain probability of being sequenced. When miRDeep maps the sequenced RNAs (‘reads’) to the genome and to the corresponding predicted miRNA precursor hairpin structure, read sequences map to the positions reminiscent of the three Dicer products. However, the mature sequences are generally more abundant in the cell and are therefore also sequenced more frequently than the loop and star sequence RNAs. Thus, the statistics of the read positions and frequencies of the reads within the stable hairpin(the ‘signature’) are highly characteristic for miRNAs and are scored by miRDeep. The power of miRNA discovery by miRDeep is proportional to the depth of sequencing. (b) Large numbers of hairpins that are not processed by Dicer are also transcribed from metazoan genomes. These hairpins can also produce short RNAs, either through non-Dicer processing or through degradation. However, when the reads that originate from such sources are mapped back to the secondary structure, they will likely map in a manner that is inconsistent with Dicer processing.
An overview of the miRDeep algorithm is shown in Figure 5. Briefly, after the sequencing reads are aligned to the genome, the algorithm excises genomic DNA bracketing these alignments and computes their secondary RNA structure. Plausible miRNA precursor sequences are then identified and, in the core part of the miRDeep algorithm, scored for their likelihood to be real miRNA precursors. The output is therefore a scored list of known and novel miRNA precursors and mature miRNAs in the deep-sequencing sample, as well as estimates for the number of false positives.
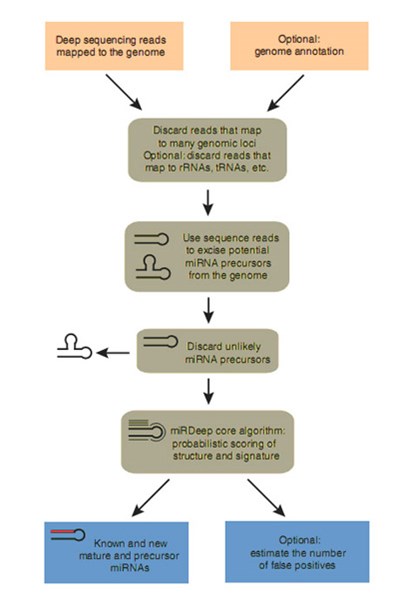
Fig 5. Flowchart diagram representing the miRDeep software package. After the sequencing reads are aligned to the genome, the algorithm excises genomic DNA bracketing these alignments and computes their secondary RNA structure, miRDeep initially investigates the secondary structure of each potential precursor as well as the positions of the reads that align to it. Next, a filtering step discards potential precursors that are grossly inconsistent with miRNA biogenesis. For the remaining (typically thousands of) potential precursors, miRDeep then probabilistically integrates deep-sequencing information based on a simple model for miRNA precursor processing by Dicer. If a sequence is an actual miRNA precursor that is expressed in the deep-sequencing sample, then one expects that one or more deep-sequencing reads correspond to one or more of the three products the mature miRNA sequence, the star sequence and the loop released when the precursor is cut by Dicer. If an miRNA precursor candidate is part of an actual transcript, but not a Dicer substrate, then deep-sequencing reads will not fit into this model of processing. Often, the reads will originate from staggered degradation products of stochastic lengths and positions. The miRDeep scores each potential miRNA precursor for a variety of parameters including energetic stability, positions and frequencies of reads with Dicer processing and finally gives the potential microRNA precursors.
Examples
The prediction of novel microRNA generally contains the following steps:
A. Mapping reads to the genome: The usable reads after adapter trimming are aligned to the reference genome using SOAP program to exclude possible contamination from other species. The mapping process allows at most two mismatches or two continuous gaps for aligning a read onto the reference sequence. The unmapped sequences are discared as unusable reads.
After mapping all the reads to the reference genome, diagrams (Fig. 6) will be given to show the percentage of reads mapped to the genome.
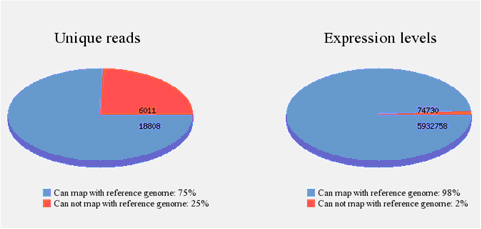
Fig 6. microRNA sequencing data from a human sample. The data is also shown in unique reads and expression levels to show the overview of genome mapping. Unique reads: sequence reads of a particular type with non-redundancy. Expression levels: All the reads from sequencing data, reflects relative abundance.
B. Classification of mapable reads: the remaining reads are then sequentially aligned to miRBase14, non-coding RNA database Rfam 9.1, repeat sequence and mRNA database, the unmapped sequences are regarded as unclassified reads for novel microRNA prediction using miRDeep.
All the reads mapped to reference genome will sequentially align to database of miRBase, Rfam, mRNA/EST and repeat sequence to identify known microRNAs and other type of RNA (Fig. 7).
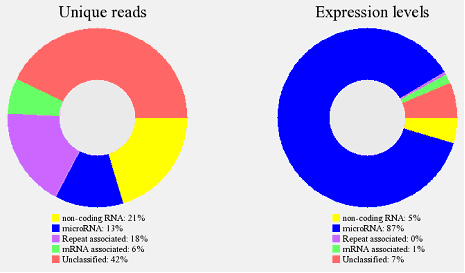
Fig 7. Proportion of miRNAs and other categories of RNA from a microRNA sequencing sample. The large-scale short reads are classified into 5 known categories. Non-coding RNA: Sequence reads mapped to Rfam 9.1; microRNA: Sequence reads aligned to miRBase 14; Repeat associated: sequence reads matched RepBase 14.09; mRNA associated: Sequence reads mapped to the human genes (UCSC, hg18); Unclassified: Sequence reads aligned to reference genome but could not be mapped to any of the above categories. The data is also shown in unique reads and expression levels to show the overview of the sequencing data. Unique reads: sequence reads of a particular type with non-redundancy. Expression levels: All the reads from sequencing data, reflects relative abundance.
C. Novel miRNA prediction: The unclassified reads will be analyzed to predict putative novel microRNA species using miRDeep. The structure and sequence information of novel microRNA candidates will be provided (Fig.8).
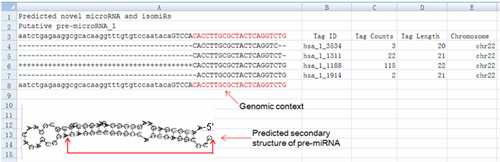
Fig 8. Novel microRNA candidates. the genome context and predicted novel microRNA and its isomiRs are shown, the respective tag counts, length and genome location are also listed. Putative fold-back structure of the pre-microRNA is given below. Tag counts: Read count number of sequences supported predicted model; Tag Length: the read length of respective sequence; Chromosome: location of predicted pre-microRNA.
Learn more about microRNA sequencing data analysis>
Reference
1. Creighton, C.J., J.G. Reid, and P.H. Gunaratne, Expression profiling of microRNAs by deep sequencing. Brief Bioinform, 2009. 10(5): p. 490-7.
2. Morin, R.D., et al., Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res, 2008. 18(4): p. 610-21.
3. Glazov, E.A., et al., A microRNA catalog of the developing chicken embryo identified by a deep sequencing approach. Genome Res, 2008. 18(6): p. 957-64.
4. Jagadeeswaran, G., et al., Deep sequencing of small RNA libraries reveals dynamic regulation of conserved and novel microRNAs and microRNA-stars during silkworm development. BMC Genomics, 2010. 11: p. 52.
5. Friedlander, M.R., et al., Discovering microRNAs from deep sequencing data using miRDeep. Nat Biotechnol, 2008. 26(4): p. 407-15.