
The Challenges and Solutions of Small RNA Profiling
The Biases of Using TPM in Small RNA Sequencing Data Analysis
Simultaneously Profile Multiple Small RNA Classes Accurately
Direct End-labeling to Avoid the Biases From Sequencing Library Prep
The comparison of small RNA transcript levels between samples using TPM in small RNA-sequencing (small RNA-seq) is actually meaningless.
In small RNA-seq, Transcripts Per Million (TPM) is the read counts of a small RNA normalized to 1 million read counts for all small RNAs in the sequenced sample, as given in the formula (1). Because the short small RNA lengths can be fully covered by single reads, the number of reads is equal to the number of small RNA transcripts. Transcript length normalization like RPKM is not needed, as given in the formula (2). TPM has been commonly used to represent relative RNA abundance levels and to compare differential expression among samples.

However, as illustrated in Fig. 1, a change in one RNA abundance level can adjust the TPM values for all other RNAs, even though the actual absolute abundance levels of the other RNAs are not changed. Therefore, TPM is dependent on the composition of the RNA population in a sample. A few very highly expressed genes can skew the distribution of TPM expression values. In fact, small RNA repertoires do change substantially under many experimental conditions or across datasets in different studies, compromising TPM to compare small RNA levels between/among samples [1,2].
Furthermore, TPMs represents relative abundance. A direct comparison of TPM between samples is meaningful only when the total RNAs are equal between the samples and the distribution of RNA populations are close to each other. However, the assumption that cells of different types or under different conditions produce similar levels of RNA is not always valid. Depending on severity, under these conditions, the direct comparison of TPM across samples may not be meaningful [1].
Therefore, using TPM as a universal quantification measure of small RNA abundances or for differential analysis can be misleading or may even become pointless. Finding a good measure to well represent small RNA-seq abundance levels is not straight-forward, and the best way to do this is still a matter of research [4].
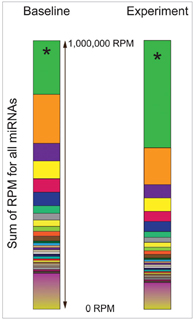
Figure 1. In this simulated situation of miRNA-seq results, except for the *RNA, all other miRNAs between Baseline and Experiment conditions are unchanged. However, the increase of only one *RNA abundance by 2-fold under Experiment condition will simultaneously depress the TPMs for all other miRNAs, even though their actual absolute expression levels are not changed [3].
Relative Service
Arraystar Small RNA Array Service
References
[1] Zhao S. et al (2020) “Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols” RNA 26(8):903-909 [PMID:32284352]
[2] Zhao Y. et al (2021) TPM, FPKM, or Normalized Counts? A Comparative Study of Quantification Measures for the Analysis of RNA-seq Data from the NCI Patient-Derived Models Repository. J Transl Med 19(1):269 [PMID:34158060]
[3] Witwer KW. and Halushka MK. (2016) Toward the promise of microRNAs – Enhancing reproducibility and rigor in microRNA research. RNA Biol 13(11):1103-1116 [PMID:27645402]
[4] Chen Y., et al (2021) “edgeR: differential analysis of sequence read count data” [https://www.bioconductor.org/packages/release/bioc/vignettes/edgeR/inst/doc/edgeRUsersGuide.pdf]