
The Challenges and Solutions of Small RNA Profiling
The Biases of Using TPM in Small RNA Sequencing Data Analysis
Simultaneously Profile Multiple Small RNA Classes Accurately
Direct End-labeling to Avoid the Biases From Sequencing Library Prep
Small RNA-seq has been used for profiling microRNAs and other small RNAs. However, the use of small RNA-seq data to quantify the relative abundances of small RNAs has been inconsistent with qPCR and Northern Blot results. For example, the level of miR-143 was measured 40 times higher than miR-145 by small RNA-seq, yet their expression levels were equivalent as measured by qPCR or Northern Blot [1-3]. The inaccuracy by small RNA-seq is primarily due to the biases in small RNA-seq library preparation and data analysis [4, 5], which can lead to compromised or misleading results, in some cases by as much as 10,000-fold off the true abundance [6-11].
The challenges of small RNA profiling
• Interference from RNA modifications
Modifications within small RNAs drastically interfere with the cDNA synthesis during the sequencing library construction and the quantification of small RNAs. Small RNA modifications (e.g. m1A, m3C and m1G) block reverse transcription and abort cDNA copying, causing sequence biases toward the priming end and failures in building unskewed full length representation in the library. For example, due to the presence of m1A modification in the TUC loop region, small RNA-seq often identifies 18-nt 3’tsRNAs but misses the more predominant 22-nt isoforms as detected by Northern Blot.
• Library amplification biases
To produce sufficient cDNA amount for sequencing instrument loading, small RNA-seq requires PCR amplification of the library [12]. However, RNA G/C content, sequence contexts, secondary structures, RNA lengths and priming, and reaction conditions can lead to biases and distortions in the PCR products. That is, when different templates in the transcriptome are amplified together in the PCR reaction, preferential or refractory amplifications for the templates invariably lead to the loss of faithful representation of RNA abundances [13]. Therefore, quantification by small RNA-seq, which requires multiple PCR amplification rounds, is never absolute and necessitates the use of an orthogonal method for validation.
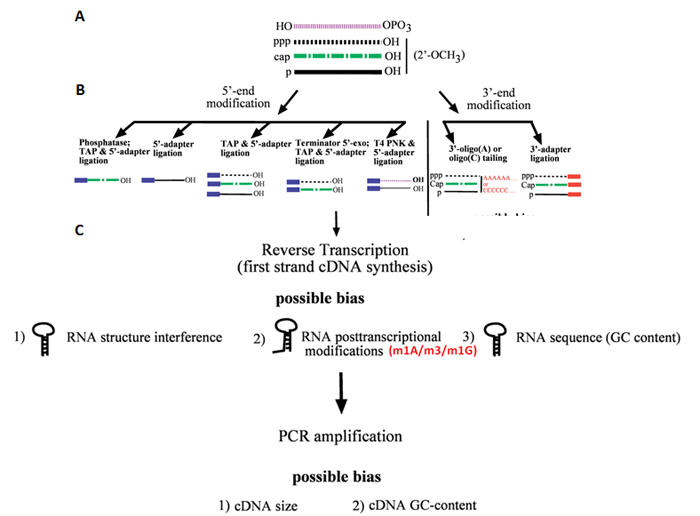
Figure 1. Illustration of the steps involved in cDNA construction, including potential sources of bias. (A) The starting pool of non-protein coding RNAs with different 50- and 30-end modifications schematically indicated by different line types. (B) The left panel depicts different enzymatic pre-treatments prior to RNA 50-end ligation to enrich for different RNA subtypes. RNA classes accessible for adapter ligation after the respective 50-end pretreatments are schematically represented below each pretreatment. The right panel depicts subtypes of RNA classes accessible for 30-end tailing (-oligo(A) or –oligo(C) tailing) and adapter ligation. (C) Possible biases associated with RNA 50-(left) and 30-(right) end-modifications and with the subsequent steps of cDNA construction.
• Caveat of TPM as the measure of abundance
Reads Per Million reads (TPM) is commonly used in small RNA-seq to represent the relative abundance of a small RNA transcript simply normalized to 1 million small RNAs being sequenced. However, a change in the read count of one small RNA will adjust the TPM values of all the other small RNAs in that total 1 million reads, even their actual absolute expression levels are not changed. Learn more>>
• Sample amounts required
For tRNA and tsRNA sequencing, 5ug, even more[14] total RNA is required for target RNA isolation and pretreatments prior to library construction, which precludes research projects with limited sample amount availability.
The solutions of small RNA profiling
Arraystar Small RNA Array is designed as a practical and effective solution to overcome these challenges to profile the full spectrum of small RNAs at high sensitivity and accuracy yet at much less input RNA amounts. Arraystar small RNA Array combines direct end-labeling and smart probe design microarray technologies to simultaneously detect and quantify small RNAs including miRNAs, pre-miRNAs, tsRNAs, tRNAs and snoRNAs on the same array (Fig. 2), providing vital expressional information to study the regulatory functions and biomarker potentials of the small RNAs.
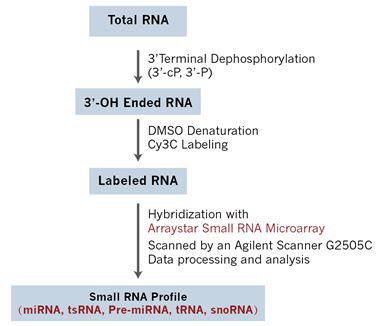
Figure 2. Workflow of Arraystar Small RNA Array Profiling. Total RNA is dephosphorylated with T4 polynucleotide kinase (T4 PNK) to remove phosphate (P) or cyclic phosphate (cP) group from the 3′ end of the small RNA to form 3-OH end. The 3-OH ended small RNAs are then denatured by DMSO and enzymatically ligated with Cy3 label. The labeled RNAs are hybridized onto an Arraystar Small RNA Microarray analyzed for small RNA expression.
Relative Service
Arraystar Small RNA Array Service
References
[1] Kent OA. et al (2010) “Repression of the miR-143/145 cluster by oncogenic Ras initiates a tumor-promoting feed-forward pathway” Genes Dev 24(24):2754-9 [PMID:21159816]
[2] Chivukula RR. et al (2014) “An essential mesenchymal function for miR-143/145 in intestinal epithelial regeneration” Cell 157(5):1104-16 [PMID:24855947]
[3] Akao Y. et al (2007) “Downregulation of microRNAs-143 and -145 in B-cell malignancies” Cancer Sci 98(12):1914-20 [PMID:17892514]
[4] Raabe, C. A., et al. (2014) “Biases in small RNA deep sequencing data” Nucleic Acids Res 42(3):1414-26 [PMID: 24198247]
[5] Witwer, K. W. and Halushka, M. K. (2016) “Toward the promise of microRNAs – Enhancing reproducibility and rigor in microRNA research” RNA Biol 13(11):1103-1116 [PMID: 27645402]
[6] McCormick, K. P., et al. (2011) “Experimental design, preprocessing, normalization and differential expression analysis of small RNA sequencing experiments” Silence 2(1):2 [PMID: 21356093]
[7] Linsen, S. E., et al. (2009) “Limitations and possibilities of small RNA digital gene expression profiling” Nat Methods 6(7):474-6 [PMID: 19564845]
[8] Sorefan, K., et al. (2012) “Reducing ligation bias of small RNAs in libraries for next generation sequencing” Silence 3(1):4 [PMID: 22647250]
[9] Hafner, M., et al. (2011) “RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries” RNA 17(9):1697-712 [PMID: 21775473]
[10] Tian, G., et al. (2010) “Sequencing bias: comparison of different protocols of microRNA library construction” BMC Biotechnol 10:64 [PMID: 20815927]
[11] Fuchs RT. et al (2015) “Bias in ligation-based small RNA sequencing library construction is determined by adaptor and RNA structure” PLoS One 10(5):e0126049 [PMID:25942392]
[12] Kapranov P. et al (2012) “Profiling of short RNAs using Helicos single-molecule sequencing” Methods Mol Biol 822:219-32 [PMID:22144202]
[13] Dabney J. and Meyer M. (2012) “Length and GC-biases during sequencing library amplification: a comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries” Biotechniques 52(2):87-94 [PMID:22313406]
[14] Junchao S., et al. (2021) “PANDORA-seq expands the repertoire of regulatory small RNAs by overcoming RNA modifications” Nat Cell Biol 23(6):676 [PMID: 33927371]