
Microarrays have been used for decades as the standard technology platform for gene expression profiling. In recent years, Next Generation Sequencing (NGS), which measures gene expression levels by quantifying transcript read densities in the whole transcriptome, has become increasingly popular for gene expression studies. Long non-coding RNAs (LncRNAs) are non-protein coding transcripts longer than 200 nt. Because of their functional roles in diverse biological processes and human diseases, LncRNAs have been a subject of active scientific research. For LncRNA gene expression profiling, microarray technology has many important and irreplaceable advantages over RNA-Seq and continues to be the platform of choice (Table 1).
LncRNAs are expressed at lower levels than protein-coding RNAs
Low abundance transcripts are not reliably quantified by RNA-Seq
Increased read depths have limited gains in accuracy for low abundance transcripts
Microarrays are more adept for low abundance LncRNA profiling
LncRNAs are comprehensively covered by microarrays
Microarray technology is well established and mature.
LncRNAs are expressed at lower levels than protein-coding RNAs
As one of their general properties, LncRNAs are poorly expressed [1-5]. Based on FPKM (fragments per kilobase per million mapped reads) values, LncRNAs are expressed at levels approximately 1/10 that of protein-coding genes (Figure 1 and Figure 2) [4-7].
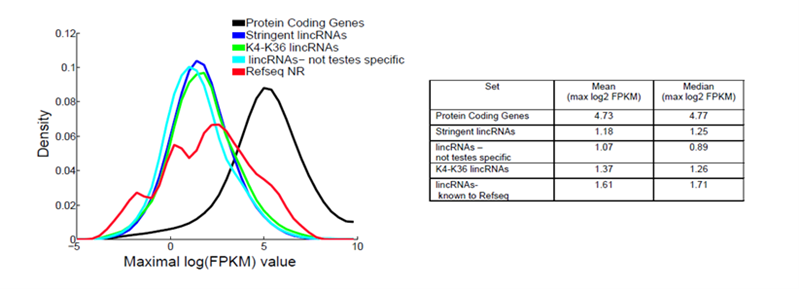
Figure 1. Density distributions of maximal expression abundance across all tissues. The log2 normalized FPKM counts are estimated by Cufflinks, showing stringent lincRNAs (blue), stringent lincRNAs with K4-K36 support (green), stringent lincRNAs which are not testes-specific (cyan), lincRNAs known to RefSeq NR (red) and protein-coding genes(black). The mean and median of maximal expression scores are presented in the table on the right [4]. The fold difference between the protein coding genes and lncRNAs can be converted from the difference of their log2FPKMs as 2Dlog2(FPKM).
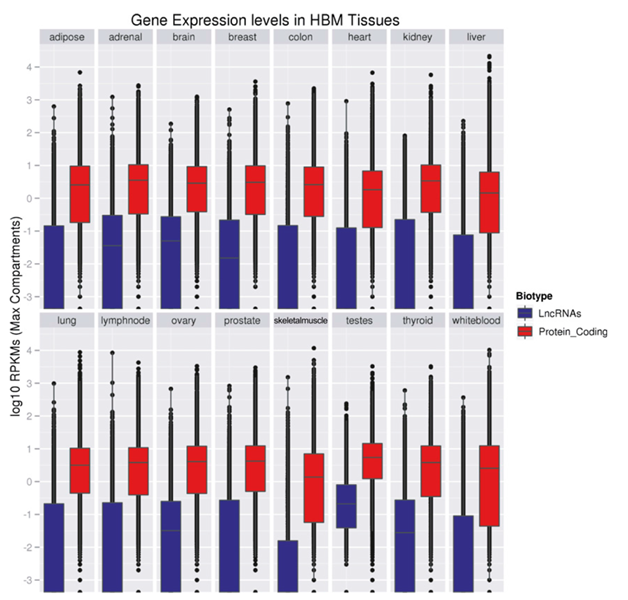
Figure 2. Distributions of LncRNA (blue) and protein-coding (red) transcripts’ expression in human tissues [7]. The vertical axis for expression level is indicated in log10 scale.
Low abundance transcripts are not reliably quantified by RNA-Seq
For low-abundance RNAs, Poisson sampling noise due to finite read depth is the dominant source of error in RNA-Seq [8]. Consequently, poorly expressed transcripts cannot be measured by RNA-Seq reliably and need enrichment [8].
The relationship between gene expression level and measurement precision was studied and characterized in datasets using three technical replicates containing 331 million 50bp reads each [9]. As seen in Figure 3, the higher the gene expression level, the more precise the measurement. Conversely, the lower the gene expression level, the higher the relative error in the measurement. As LncRNAs are expressed at levels much lower than that of protein coding genes, a signification proportion of the lower abundance LncRNAs cannot be precisely quantified for their expression by RNA-seq (relative error greater than 20%).
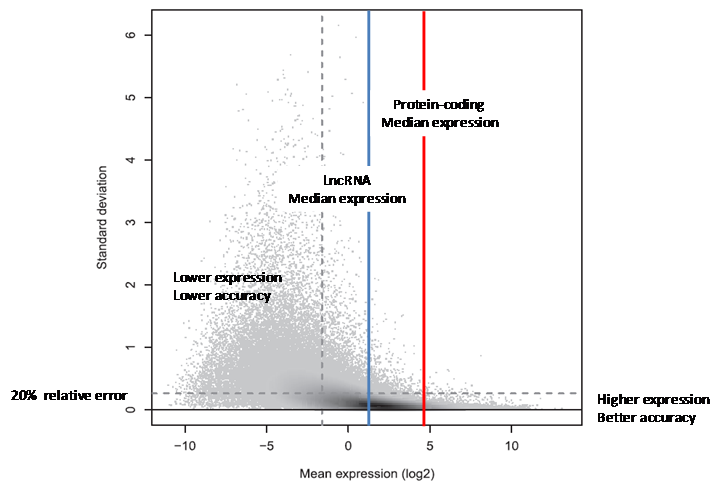
Figure 3. Standard deviation versus expression level. The plot shows the variation across three technical replicate measurements (standard deviation, y-axis), with each discernible dot representing a transcript target. In the shaded areas, the grey level represents density, with dark shading indicating higher densities. The standard deviation is in general larger for transcripts with lower mean expression levels (x-axis). More strongly expressed transcripts could often be measured reliably, with a relative error of 20% or less. Interestingly, just 41% of all transcript targets could be measured with such precision (below the horizontal dashed line). Of the 41% most strongly expressed transcripts (to the right of the vertical dashed line), on the other hand, 84% could be measured reliably (below the horizontal dashed line). This is reflected by the high density of targets on the right (dark shading) falling largely below the horizontal line, which is not the case to the left of the vertical dashed line. (Reproduced from [9]).
Increased read depths have limited gains in accuracy for low abundance transcripts
In order to measure the expression levels more accurately, one could increase the RNA-seq read depths. Generally, 100 million reads are sufficient to detect most expressed genes and transcripts, but 500 million reads are needed to quantify the majority (i.e. 72%) of the gene expression levels accurately [10]. However, increased read depths only have a significant impact on the accuracy of abundantly-expressed transcripts, whereas the accuracy of poorly-expressed transcripts is relatively unchanged with increasing read depths, as seen in the case studies of PHB, CD74, and BRD4 transcript isoforms at different abundance levels [10].
Overall, an unlimited increase in the number of RNA-seq reads will not result in an unlimited increase in the number of low abundance transcripts whose expression levels can be reliably quantified. Increased read depths have diminishing gains that will be maxed out (Figure 4) [9]. In RNA-Seq, 7% of the abundant transcripts dominate over 75% of all read alignments. Most of the added measurement power is wasted on a small number of abundant transcripts, such as housekeeping genes. This implies that the expression levels of certain low abundance LncRNAs cannot be measured precisely at practical read depths or even at all despite the ultra-deep reads.
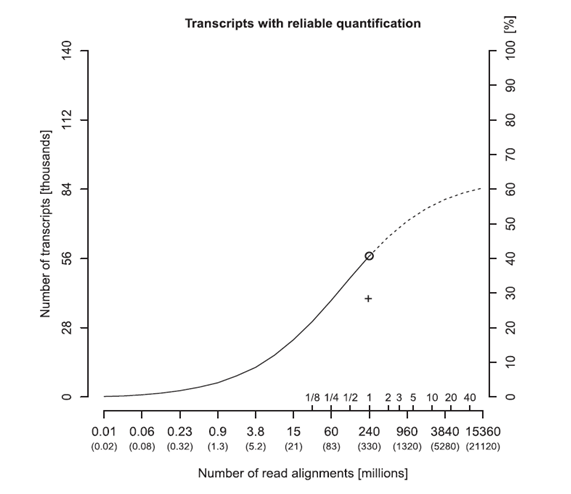
Figure 4. Transcripts with reliable quantification with a relative error of 20% or less versus read depth. The total number of generated reads is given in parentheses below. Additional inside tick marks indicate the number of sequencing runs worth of sequencing. The alternate y-axis on the right shows the percentage of all known transcripts measured reliably. The circle symbol indicates reads (331 million reads) in an entire sequencing run. Extrapolation of the fitted sigmoid curve suggests a maximum of 60% even at reads of 10 billion.
Microarrays are more adept for low abundance LncRNA profiling
In microarray profiling, RNAs are interrogated by hybridization with sequence-specific probes. For a particular gene, the presence of unrelated sequences even in high abundance behaves no differently than carrier RNA in the hybridization solution, with little to no effect on detection of poorly-expressed transcripts. In RNA-Seq, however, the lion’s share of sequencing reads is occupied by high abundance RNAs such as housekeeping genes, leaving the low abundance RNAs poorly covered by low read depths. The low reads account for low sensitivity and reliability. Therefore, microarrays have better performance characteristics for low abundance RNAs. For example, 7000~12,000 LncRNAs are routinely detectable by microarrays vs. 1000~4000 lncRNAs by RNA-seq requiring more than 120 million reads [11]. In a clinical study, microarrays were chosen over RNA-seq because, among other considerations, the better sensitivity [12].
LncRNAs are comprehensively covered by microarrays
One of the enticing capabilities of RNA-seq is to discover new and novel transcripts. Unlike microarrays that only measure the expression of known sequences, RNA-seq operates on both known and unknown sequences. Nevertheless, with the accumulation of human expressed sequences already ongoing for decades, novel transcripts discovered by RNA-seq have become few and far between [10]. More often, RNA-seq library preparation artifacts and immature RNAs are mistakenly reported as novel transcripts. With advanced microarray designs, transcript variants are routinely distinguished by exon-junction specific probes in the microarrays. Thus, investigators using microarrays, particularly those with the most up-to-date LncRNA collections and with transcript variant specific designs, are not missing out.
Microarray technology is well established and mature.
The current consensus is that “RNA-seq is not a mature technology, which is undergoing rapid evolution of biochemistry of sample preparation; of sequencing platforms; of computational pipelines; and of subsequent analysis methods that include statistical treatments and transcript model building” (The ENCODE Consortium) [13].
Table 1. Comparisons of Microarray and RNA-Seq in lncRNA expression profiling
| Microarray | RNA-Seq |
| Low abundance RNAs are interrogated by hybridization with the sequence specific probes, independent of high abundance RNAs. | The lower the gene expression level, the poorer the accuracy. Low abundance RNA reads are displaced by high abundance RNAs, making the quantification of low abundance RNAs unreliable. |
| Better sensitivity and accuracy for low abundance RNAs such as LncRNAs [12]. For example, 7000~12,000 LncRNAs are routinely detectable by microarray. | Less sensitive for low abundance RNAs. 1000~4000 LncRNAs detectable by RNA-seq with sequencing reads at 120 million [11]. |
| Strand specific detection of antisense LncRNAs, a very important group of lncRNAs. 50~70% of all transcription units in the genome contain antisense transcripts. | No. |
| Microarray processing steps, such as sample labeling and hybridization, can be carried out for many samples concurrently. Higher sample processing throughput is important for studies involving large sample sizes or research projects involving many data points [12]. | Most next-generation sequencing systems can only handle one or a few samples at a time, tying up extended periods of system time. |
| Simple and reliable aRNA synthesis/labeling reaction. | Trickier NGS library preparations. |
| Less intensive in computational resources and less demanding in bioinformatics support. | Computationally intensive. Bulky data storage. Demanding in bioinformatics support. |
| Most up-to-date LncRNA collections and transcript variant specific designs have the LncRNAs covered. | Novel transcript detection. However, RNA-seq library preparation artifacts and immature RNAs are mistaken as novel transcripts. With the accumulation of human expressed sequences already ongoing for decades, novel transcripts discovered by RNA-seq have become few and far between. |
| Mature and established technology | RNA-seq is not a mature technology, which is undergoing rapid evolution of biochemistry of sample preparation; of sequencing platforms; of computational pipelines; and of subsequent analysis methods that include statistical treatments and transcript model building [13]. |
Related Services
LncRNA Array Service
SE-lncRNA Array Service
LncPath™ Array Service
T-UCR Array Service
References
1. Kampa, D., et al., Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res, 2004. 14(3): p. 331-42.
2. Cawley, S., et al., Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell, 2004. 116(4): p. 499-509.
3. Ravasi, T., et al., Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res, 2006. 16(1): p. 11-9.
4. Cabili, M.N., et al., Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev, 2011. 25(18): p. 1915-27.
5. Guttman, M., et al., Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol, 2010. 28(5): p. 503-10.
6. Yan, L., et al., Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol, 2013.
7. Derrien, T., et al., The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res, 2012. 22(9): p. 1775-89.
8. Jiang, L., et al., Synthetic spike-in standards for RNA-seq experiments. Genome Res, 2011. 21(9): p. 1543-51.
9. Labaj, P.P., et al., Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics, 2011. 27(13): p. i383-91.
10. Toung, J.M., et al., RNA-sequence analysis of human B-cells. Genome Res, 2011. 21(6): p. 991-8.
11. Kretz, M., et al., Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev, 2012. 26(4): p. 338-43.
12. Xu, W., et al., Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A, 2011. 108(9): p. 3707-12.
13. The_ENCODE_Consortium, Standards, Guidelines and Best Practices for RNASeq. http://encodeproject.org/ENCODE/protocols/dataStandards/ENCODE_RNAseq_Standards_V1.0.pdf, 2011.